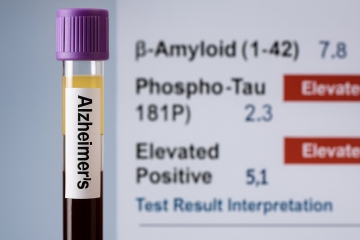

알츠하이머 치료제 \'르켐비(Leqembi)\'의 정체된 시장 반응 속, 미국 식품의약국(FDA)이 새로운 판을 열 수 있는 진단 도구를 승인했다. 지난 5월 17일, FDA는 후지레비오 다이애그노스틱스(Fujirebio Diagnostics)의 루미펄스(Lumipulse) 혈액검사를 정식 승인하며, 알츠하이머 질환 진단 경로의 간소화 가능성에 신호탄을 쏘아올렸다.
루미펄스는 혈장에서 측정한 pTau217과 아밀로이드 β1-42의 비율을 바탕으로, 뇌 속 아밀로이드 플라크 축적 여부를 조기 감지할 수 있도록 설계됐다. 이는 르켐비 및 일라이 릴리의 신약 ‘키순라(Kisunla)’가 타깃하는 주요 병리기전이기도 하다.
기존에는 르켐비 투약 전, PET 스캔이나 뇌척수액(CSF) 검사를 통해 아밀로이드 플라크 존재 여부를 반드시 확인해야 했다. 하지만 PET은 고가이면서 의료 인프라 의존도가 높고, CSF 검사는 침습적이라는 한계가 있었다. 이런 현실 속에서 루미펄스는 ‘진입장벽을 낮추는 첫 열쇠’로 평가받고 있다.
루미펄스는 단독 진단도구로 사용되지는 않으며, 임상정보와 병합 평가가 필요하다는 FDA의 제한 조건이 붙었다. 그러나 Mizuho의 애널리스트 살림 사이드는 해당 검사가 PET이나 CSF 검사 전단계에서 \'걸러내기(rule-out)\' 용도로 활용될 경우, 전체 진단 흐름의 효율성을 높일 수 있다고 분석했다.
루미펄스의 예측력도 주목된다. 양성 예측값은 91.7%, 음성 예측값은 97.3%로, 위양성 및 위음성 가능성을 줄인다는 점에서 신뢰성이 확보됐다. 특히 음성 결과에 대한 신뢰도가 높기 때문에, 고위험군을 선별하고 PET 검사의 과잉 사용을 줄이는 전략적 도구가 될 수 있다.
현재 진료 현장에서 알츠하이머 치료 후보자로 검토되는 환자 중 절반가량은 병이 너무 진행되어 있어 르켐비 투약 대상이 되지 않는다. 바이오젠 CEO 크리스 비에바허는 “혈액검사를 통해 조기 선별이 가능해진다면, 신경과 진료 현장에서 치료 적합 환자 비율을 끌어올릴 수 있다”고 밝혔다.
다만 전문가들은 루미펄스의 임상 적용을 위해서는 여전히 ‘교육’이 필요하다고 지적한다. 단순한 승인만으로는 의사의 처방 행태 변화가 어려우며, 특히 기존의 PET 및 CSF 검사와의 임상적 연계 해석이 관건이라는 것이다.
루미펄스는 FDA 승인 이전에도 몇몇 ‘실험실 개발 검사(LDT)’ 형태로 혈액기반 진단이 존재했지만, 이들은 신뢰 부족으로 실제 사용률이 저조했다. 반면 루미펄스는 FDA의 정식 허가와 90% 이상의 정확도를 기반으로, 신경과 전문의를 비롯한 의료진의 신뢰를 끌어낼 수 있을 것으로 기대된다.
그러나 루미펄스가 르켐비 처방 확대에 실질적 영향을 미치기 위해서는 ‘보험 급여’ 확보가 핵심 변수다. 현재로서는 CMS(미국 메디케어·메디케이드 서비스센터)의 고급 진단 검사(ADLT) 지정을 통해 별도 급여 코드를 받을 수 있어야 비용 측면에서 환자 접근성이 열릴 수 있다.
FDA 승인 외에도, 르켐비의 미래 성장은 혈액검사 보급과 함께 ‘피하주사형 제형’의 상용화 여부에 달려 있다. 에자이와 바이오젠은 이 두 요소를 르켐비의 장기적 성장 축으로 보고 있으며, 실제 미국 매출은 2025년 3월까지 약 171백만 달러를 기록했고, 내년에는 400억 엔(약 2억6천만 달러) 이상을 목표로 잡고 있다.